第一作者:袁建华
通讯作者:马杰教授,曹江林教授
通讯单位:同济大学
论文DOI:10.1016/j.nantod.2025.102781

研究背景
LFP作为Li+捕获阴极的一个关键障碍是开发经济的制造工艺。共沉淀法、水热法、微波转化等低温合成方法为制造这种电极材料提供了可操作、低成本以及高效率的途径,然而,不同低温合成方法导致的反位缺陷(Li和Fe)仍是当前合成LFP的一个主要问题。LFP中的Li+运动是通过一维(1D)通道(沿b轴)进行,反位缺陷不仅会阻碍Li+沿最快的扩散路径传输,而且还会引起额外的静电排斥,从而导致LFP晶体结构不稳定。目前,合成具有低浓度反位缺陷的LFP可有效改善其电化学性能已被多位学者证实。因此,开发绿色经济并含有低浓度反位缺陷的LFP合成策略对于改善扩散动力学及循环稳定性具有重要意义。
微生物“代谢催化”是一种具有吸引力的小尺寸材料合成方法,通常在常温、常压等温和条件下进行,因此投资少、能耗低且安全性高。“代谢催化”过程是由氧化还原酶、异构酶、裂合酶和合成酶等多种酶共同作用的结果,这些酶高度专一,催化效率比常规化学催化反应高107~1013倍,从而保证了目标产物的快速、可控合成。微生物代谢催化是构建空位的有效策略之一。这一过程涉及微生物通过新陈代谢和组装将金属离子转化为含有缺陷的无机化合物。微生物的转运蛋白和氧化还原酶等活性分子可加速产物的转化和合成。通过微生物新陈代谢引入缺陷的研究现已取得阶段性进展。此外,“代谢催化”在合成材料的稳定性方面具有独特优势,微生物分泌的酶可作为还原剂、封盖剂和稳定剂等,通过代谢和组装提高合成产物的稳定性。已有研究通过使用还原细菌,如希瓦氏菌属(Haejae-1),将Fe3+还原为Fe2+,从而在常温厌氧条件下合成LFP,但这种厌氧细菌培养困难、合成条件苛刻、合成周期长,并未表现出独特的优势。此外,目前关于利用微生物“代谢催化”功能制备低浓度缺陷LFP的研究尚无,同时如何进一步提高LFP的Li+选择性能仍面临严峻挑战。
基于上述考虑,进一步筛选适配微生物-聚磷菌,利用其“代谢催化”功能直接合成了LFP@C电极材料。LFP@C具有多种优势,如低浓度反位缺陷LFP提供了通畅的离子扩散通道,促进了Li+扩散和电子传输,从而有效地改善了LFP的动力学,同时其稳定的晶体形态可有效承受Li+插入和脱除引起的体积变化,从而提高了电极的循环稳定性;得益于这些优势,LFP@C表现出优异的Li+捕获容量、速率、高选择系数以及良好的循环稳定性。本章节创新引入了一种绿色合成低浓度反位缺陷LFP的新方法。
内容简介
通过筛选适配微生物-聚磷菌,利用其“代谢催化”功能直接合成电极材料。基于聚磷菌自身所含酶类将摄入离子还原、转化、封盖和稳定的特性,实现PO43-、Fe3+和Li+的“代谢催化”转化,进一步热解获得磷酸铁锂与衍生碳复合的电极材料LFP@C。考察pH对合成产物形貌的影响,在不同pH条件下,LFP的形貌分别为绣球状(pH= 5)、手风琴(pH= 6)、海胆状(pH = 7)和海参状(pH = 8)。LFP@C-7的尺寸为50-100 nm,比表面积为200.8 m2g–1,LFP@C-7中LFP的含量约为83.2%。作为Li+选择性阴极时,LFP@C-7的Li+分离容量为3.05 mmol g-1,明显高于Ca2+(0.89 mmol g−1)和Mg2+(1.06 mmol g−1),当Mg2+/Li+比为60时,选择性系数达到峰值(212),表明其具有优异的Li+选择性能,具备从高Mg2+浓度盐水中分离Li+的潜力。X射线衍射、拉曼、红外测试发现LFP@C含有低浓度反位缺陷(< 3%)和稳定的晶体形态,从而有利于Li+沿一维通道的快速传输及保持LFP稳定的橄榄石结构。电化学测试发现LFP@C-7较其他样品具有更大的比电容(250 F g-1)和离子扩散系数(8.20×10-14cm2s-1),同时仅在LiCl溶液中出现充放电反应平台和氧化还原峰,而在其他溶液(CaCl2、KCl、MgCl和NaCl)中均未出现,进一步证实其具有选择性分离Li+的优势。EQCM-D测试发现Li⁺先发生水合,接着在LFP层间完成水化层脱除,该过程提高了Li⁺层间迁移速率并增加了活性面积,从而增强了Li⁺分离性能,分离阶段的质量守恒证实Li⁺脱嵌过程的可逆。原位XRD结果发现Li+脱嵌过程发生了FePO4→LiFePO4→FePO4的转化反应,可逆相变的循环有效提升了LFP@C-7的稳定性(100次后,保持率>83.3%)。
图文导读
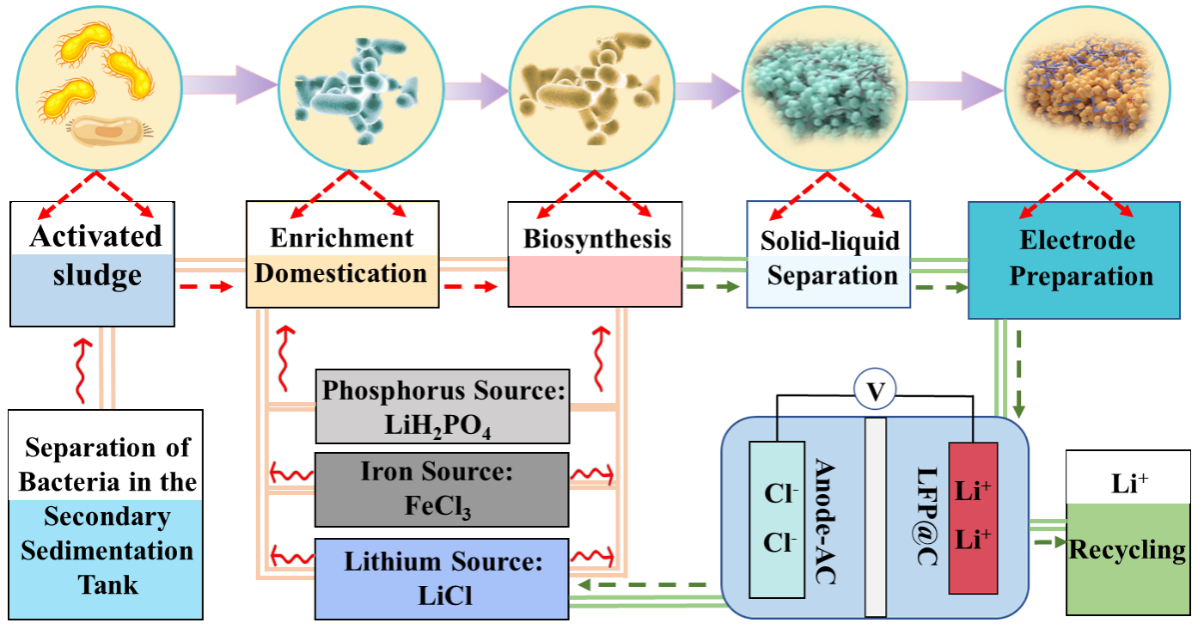
Figure 1.Schematic illustration ofBio-green synthesis of small-sized, low-concentration antisite defects LFPs for Li+recovery in a full-chain route.
图1展示了从合成到应用的全链条,即利用驯化的聚磷菌种代谢催化含低浓度反位缺陷的LFP并将其用作高效捕获Li+的CDI电极。首先,从二次沉淀池中提取活性污泥,然后进行持续驯化,培养出对PO43-、Fe3+和Li+具有较强吸附能力的混合聚磷菌种。在合成LFP时,首先活化功能性聚磷菌,随后依次加入LiH2PO4(磷源)和FeCl3(铁源)。聚磷菌将磷代谢后,引入LiCl(Li+源),通过代谢转运、代谢装配和酶催化过程合成LFP。同时,通过优化pH值范围(5-8),合成了四种形态不同的LFP,分别命名为LFP@C-5、LFP@C-6、LFP@C-7和LFP@C-8。这些样品随后被制备成电极,用于CDI系统中捕获Li+。最后,部分回收的Li+可再次作为聚磷菌合成过程中所需的锂源使用,其余部分Li+可回收存储。
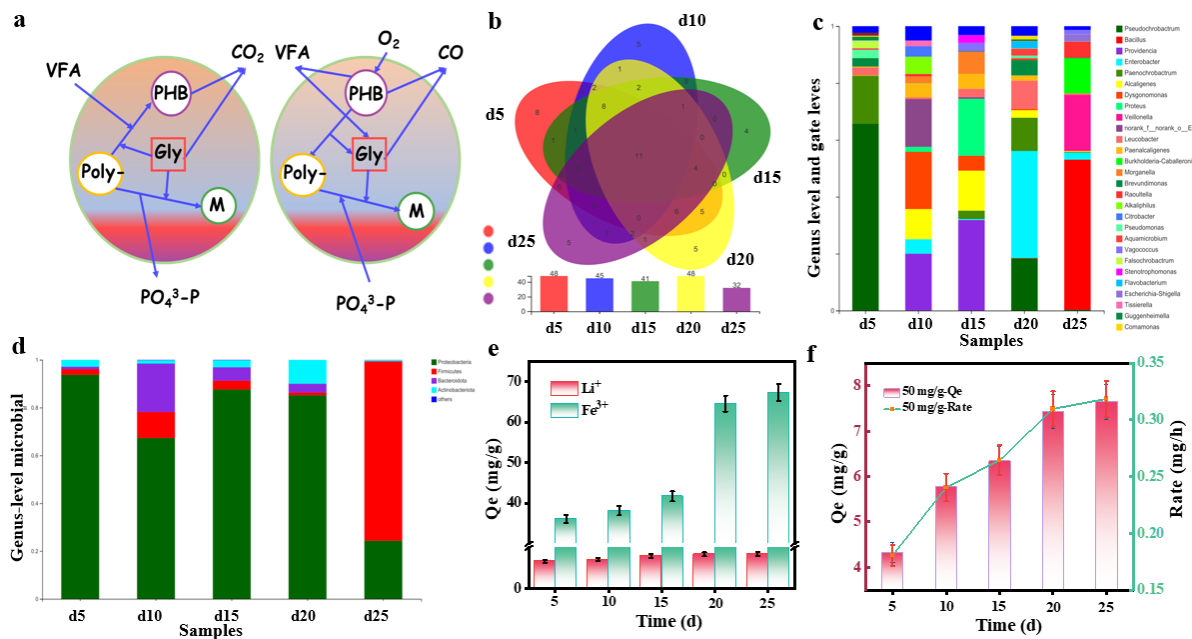
Figure 2.(a) Mechanistic diagram of phosphorus uptake and release by thepolyphosphorusbacteria. (b) Genus-level microbial community structure at different time points.Microbial community structure at the genus level (c) and phylum level (d) during different acclimation periods.(e) Changes in the adsorption capacity of strains for Fe3+and Li+. (f) Changes in capacity and rate of PO43-adsorption by strains at different stages of domestication.
进一步分析了驯化阶段聚磷菌种的微生物多样性。首先,基于维恩图分析了五个采样时间点的OTU分布差异,如图2所示,样品总OTU数为38个,五个时间点共有的OTU数为11个,占总OTU数的28.95%,且五个采样时间点的群落相似度较高,表明主要功能微生物群落在驯化过程后得到了富集。接着进行了微生物群落组成分析,5个样本在属级上共有的优势菌属是白僵菌,不同阶段的优势菌属分别占2.57%~9.88%。功能菌的比例分别为65.71%、20.08%、31.83%、37.62%和52.76%,表明不同阶段功能菌的比例存在显著差异,这可能与各菌株对PO43-、Fe3+和Li+的耐受性增强有关。最后,对微生物群落结构进行了门级分析,不同阶段的菌株在门级上共有的优势菌门包括变形菌门、固着菌门、类杆菌门和放线菌门,这表明经过多次驯化,聚磷菌群的结构逐渐稳定。
此外,对不同驯化阶段功能型聚磷菌的PO43-、Fe3+和Li+的吸附性能进行了研究,发现随着驯化时间的增长,Fe3+的吸附能力逐渐增加,最高达到68 mg g-1,而Li+的吸附能力基本保持在18 mg g-1。此外,PO43-的吸附能力随驯化时间的增长明显提高,最大吸附性能达到7.5 mg g-1,最大吸附速率达到0.32 mg h-1,这表明获得的聚磷菌具有合成LFP@C的基础,可进一步作为制备LFP的前体物质。
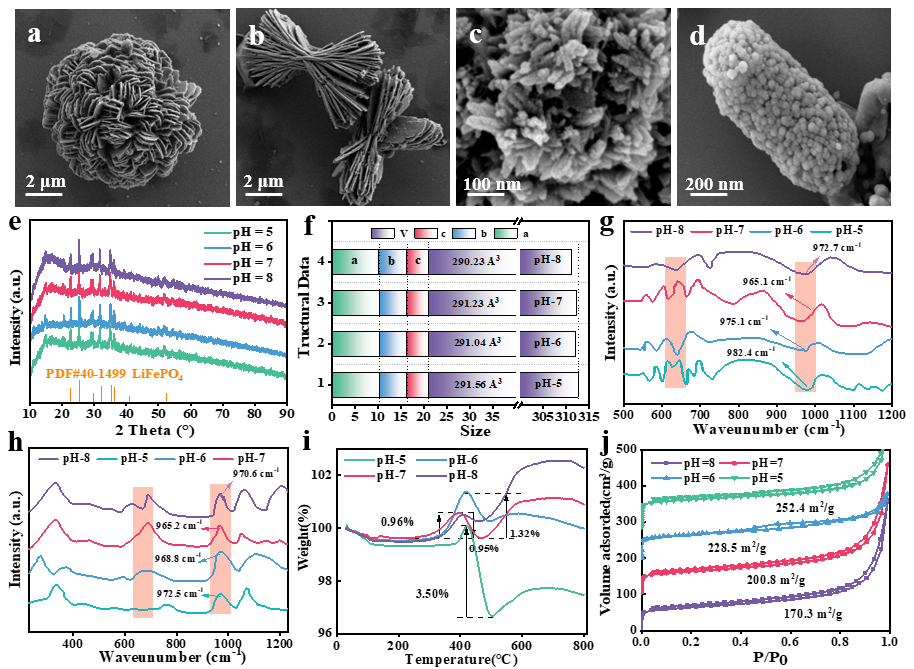
Figure 3.SEM (a: LFP@C-5, b: LFP@C-6, c: LFP@C-7, d:LFP@C-8). (e) XRD pattern of the LFP@C-7. (f)Main structural data of the LFP samples. (g)FTIR and Raman spectra (h) of LFP@C. (i) LFP Thermogravimetric tests (TGA) of LFP@C. (j)N2adsorption/desorption isotherm curves of LFP@C.
XRD结果分析显示,24~26°附近的峰值归因于菌体衍生碳。四个LFP@C样品的XRD图样的衍射峰均可被索引为LiFePO4(PDF#40-1499)。利用SEM、TEM分析了合成LFP@C的形态和结构。在不同的pH条件(pH = 5~8)下,合成了绣球花状、手风琴状、海胆状和海参状LFP。当pH小于6时,合成的LFP尺寸约为10~20μm,当pH大于6时,尺寸为20~50 nm,这可能与聚磷细菌表面的负电荷含量及不同pH条件下的离子间相互作用的大小有关。通过pH对聚磷细菌的生长繁殖和产品合成的影响,分析不同pH条件下,样品形貌和产量的差异。首先,pH值影响细菌内酶的活性,如果pH值抑制了特定酶的活性,可能会阻碍细菌的新陈代谢,从而影响产品的形态和粒径。其次,pH值影响细胞膜表面的电荷,导致膜通透性的改变,这种改变可能影响细菌的营养吸收和合成产物的释放。同时,pH会影响水中某些成分的解离,从而影响聚磷细菌对这些成分的吸收。最后,不同的pH值可诱导细菌细胞中不同的代谢过程,从而改变所产生代谢产物的质量和形貌。
如图3所示,从XRD图谱得到了LFP晶体结构的相关参数。四个样品的晶格参数(a = 10.3470 Å,b = 6.0189 Å,c = 4.7003 Å)显示出较高的结晶度,与无缺陷标准样品接近。单位晶胞的体积(V)可作为评估反位缺陷存在的关键参数。通常情况下,无缺陷的LFP的V应在290~291 Å3的范围内。经过比较发现,除了LFP@C-5的V = 291.56 Å3外,所有样品的V值均在290~291 Å3范围内。根据文献比对,LFP@C-5的反位缺陷浓度小于3%。因此,数据表明,所有代谢催化的LFP样品都可被归类为反位缺陷含量较低的样品,且反位缺陷含量均小于3%。
对所有代谢催化的LFP样品进行FTIR和拉曼表征,以进一步检查是否存在反位缺陷。FTIR中920~1150 cm-1范围内的宽带被归因于PO43-的分子间对称和非对称拉伸模式,645和527 cm-1处的峰值则源自P-O键的非对称弯曲,根据理论计算,天然P-O对称伸展振动(包括缺陷)的吸附带约为1000 cm-1,而无缺陷的LFP的吸附带则位于957 cm-1。在四种LFP的拉曼光谱结果中,957 cm-1附近的尖锐峰值对应于PO4四面体中P-O键的对称伸展,与无缺陷LFP的峰值位置一致。这一观察结果与无缺陷 LFP 的峰值位置基本相同,表明通过代谢催化的所有 LFP 都具有低浓度的反位缺陷。
TGA用于确定LFP@C中菌体衍生碳的含量。在空气条件下,LFP@C的反应发生为LiFePO4/C+O2-Li3Fe2(PO4)3+Fe2O3+CO2↑。根据文献报道,LFP的氧化产生两种氧化产物,Li3Fe2(PO4)3和Fe2O3,使体系质量增加约5.1%。因此,可计算得出LFP@C-5、LFP@C-6、LFP@C-7和LFP@C-8的碳含量分别为15.5%、10.6%、11.8%和9.50%,而LFP含量分别为79.2%、80.5%、83.2%和87.4%,其中LFP@C-5的碳含量最高,而LFP@C-8的LFP含量最低,这主要与pH差异、聚磷菌含量、细菌代谢活性以及LFP形成后的堆叠模式相关。
进一步获得了LFP@C的氮吸附-脱附吸附等温线和孔径分布。四个LFP@C样品的比表面积范围为170~252m2g-1,这是因为聚磷菌煅烧后呈中空结构,从而提高了LFP@C的比表面积。样品不同比表面积的差异可能是由不同pH条件下的细菌含量导致。比表面积增加,使LFP暴露更多活性位点,从而显著提高Li+的存储能力。带有滞后环的IV型吸附等温线和孔径分布证实了LFP@C中存在微孔和中孔。接触角测试结果表明,LFP@-7的亲疏水能力处理中间值,这可能与碳含量密切相关。
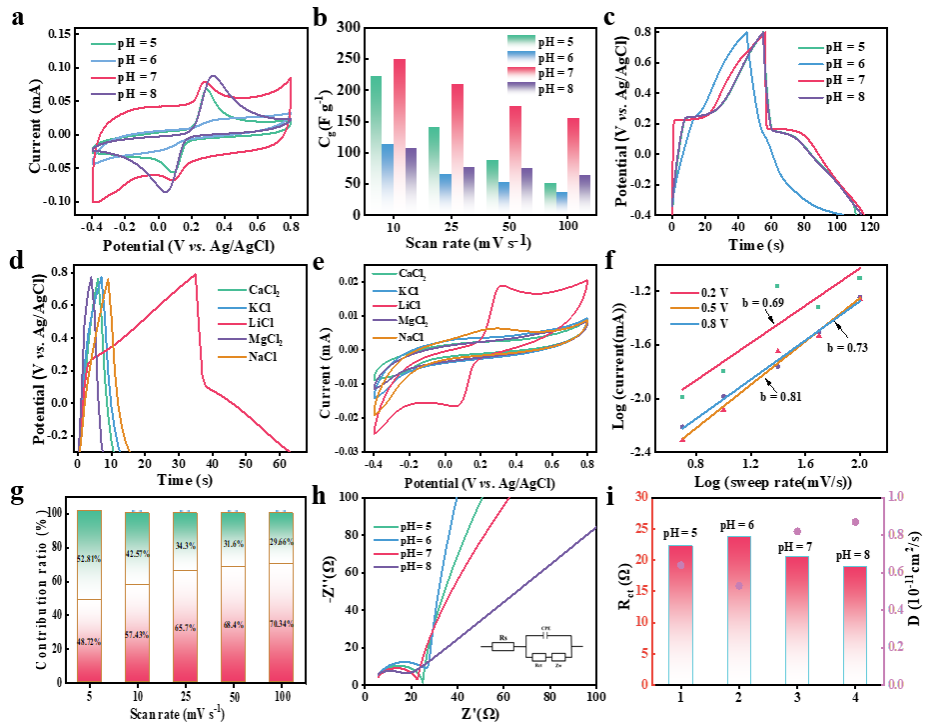
Figure 4.(a) Cyclic voltammograms for different LFP@C-7 at scan rate of 50 mV s-1. (b)Gravimetricspecific capacitances of LFP@C-7 at different scan rates. (c) GCD profiles of the LFP@C-7 at the current density of 50 mA g-1. (d) GCD profiles of LFP@C-7 in different salt solutions at the current density of 50 mA g-1.(e) CV curves of LFP@C-7 in different salt solutions at scan rate of 50 mV s-1. (f) Calculation of b-values based on CV curves.Normalized contribution ratio of capacitive capacitance at different scan rates (g). (h, i) Nyquist plots, Rctand Diffusion coefficient of different samples.
在不同的扫描速率下对所有电极的CV曲线进行了考察。四种样品均观察到明显的氧化和还原峰,其峰位置与先前研究相似。LFP@C-5、LFP@C-6、LFP@C-7和LFP@C-8对Ag/AgCl的氧化峰分别在0.282、0.322、0.277和0.322 V,表明从FePO4向LFP的转化可逆。相反,与Ag/AgCl相比,它们在0.088、0.023、0.088和0.043 V的还原峰为FP的形成提供了证据(图4)。此外,在5~100 mV s-1的扫描速度下对不同样品进行了CV扫描,发现LFP@C-7的CV积分面积最大,表明其具有更高的比电容。在25 mV s-1的扫描速率下循环100次后,发现LFP@C-7电极的CV曲线高度重叠,证明样品具有良好的循环稳定性,离子嵌入和脱除对电极的影响较小。通过计算CDI系统中的赝电容百分比为53%,EDL百分比为47%,可以看出 LFP@C-7成功地耦合了这两种机制。
通过GCD测试进一步考察了样品的电化学性能。在电流密度为50 mA g-1时,LFP@C-7的充放电时间最长,表明其具有显著的Li+捕获优势[330]。在电流密度相同的情况下,所有GCD曲线都显示出平台,这为电极在充放电周期中的赝电容行为提供了证据。相较于其他样品,LFP@C-7在不同电流密度(12.5-100 mA g-1)均显示出更高的比电容,这与CV结果高度一致。
进一步比较了LFP@C-7在1 mol L-1不同单阳离子电解质(CaCl2、KCl、LiCl、MgCl和NaCl)中的电化学性能。GCD曲线表明,LFP@C-7在LiCl溶液中的充放电时间最长,并出现反应平台,而在其他电解质溶液中充放电时间较短,且未出现反应平台,这清楚地展示了LFP@C-7的Li+选择性优势。此外,从不同电解质的CV曲线可以发现,LFP@C-7仅在LiCl溶液中出现了氧化还原峰,即氧化还原反应,而在其他电解质溶液中没有观察到氧化还原反应,这进一步表明电化学反应过程发生了Li+的嵌入和去除。
进一步计算了LFP@C-7电极的b值,根据电位为0.20 V、0.50 V和0.80 V的区域计算得出的b值介于0.69~0.81之间,表明动态过程涉及扩散和电容两个过程。在CV测试的基础上,进一步研究了Li+插层的动力学行为和电容贡献,以考察所合成样品在电化学方面的优越性。结果发现, LFP@C-7电极的电容贡献率从5 mV s-1时的48.72%增加到100 mV s-1时的 70.34%(图中粉红色为电容贡献,绿色为扩散贡献)。这表明LFP@C-7的反应主要是由电容效应控制的,因而具有较高的反应速率和循环稳定性。
此外,还利用EIS评估了样品的Li+反应动力学。所有电极的Nyquist图显示了小的半圆和较强的线性。根据EIS曲线高频段的半圆,四个样品的Rs大小基本相同。LFP@C-7的Rct(20.53 Ω)小于LFP@C-5(22.29Ω)、LFP@C-6(23.69 Ω),表明LFP@C-7由于衍生碳形成的导电骨架具有更好的电子导电性。此外,通过计算相应的D值,可以明显发现LFP@C-7的D(0.82×10-13cm2s-1)高于LFP@C-5(0.64×10-13cm2s-1)和LFP@C-6(0.53×10-13cm2s-1)。LFP@C-7电化学性能提高可能与这些方面有关。首先,LFP@-7采用 MEC策略合成,具有较低浓度的反位缺陷,其海胆状形态具有平滑的多Li+传输通道,有利于快速传输和捕获Li+,从而提高了反应动力学。同时,LFP的纳米级高结晶结构缩短了Li+沿1D通道的传输路径长度,减少了通道堵塞和电极极化。此外,LFP@-7的碳含量适中,可保持复合电极的能量密度和比电容,最后,LFP@-7具有200.8 m2g-1的高比表面积以及丰富的微孔和中孔,从而提供了充足的接触点和反应区,这些因素共同提升了LFP的电化学性能。
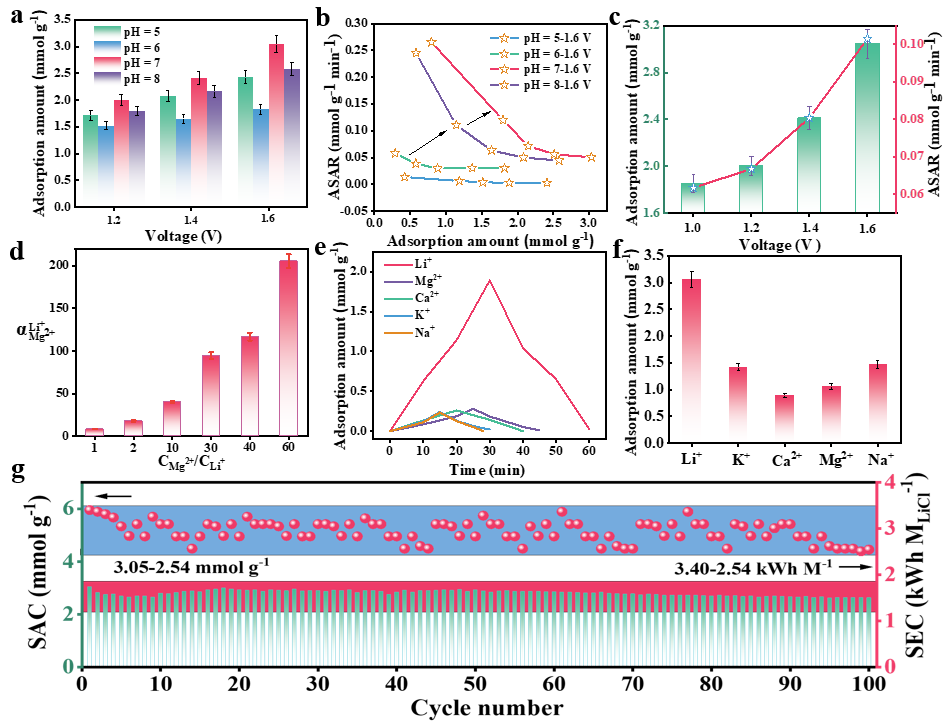
Figure 5.(a) Adsorption capacityat different voltage of different samples. (b) Kim-Yoon plots for different electrodes. (c) Adsorption capacity at different voltage of LFP@C-7. (d) The separation factor α in mixed Mg2+/Li+solutions with varying Mg2+/Li+ratios within the cell: LFP@C-7//AC. (e) The adsorption amount of thecapacitor deionization(CDI)cell performed in 10 × 10−3m single-salt solution (LiCl, NaCl, KCl, MgCl2, and CaCl2). (f)The adsorption capacity of different ions (Li+, Na+, K+, Mg2+, and Ca2+) in simulated brine water. (g)Adsorptioncapacity and SEC of CDI cell for 100 cycles at1.20 V.
图5显示了不同样品在不同截止电压(1.20~1.60 V)和1000mg L−1LiCl溶液中的Li+分离性能。在不同截止电压下,LFP@C-7的分离容量最高,其次是LFP@C-8,这与样品的形态、比表面积的大小和活性物质的含量密切相关。为了评估不同电极在1.60 V截止电压下的性能,绘制了Kim-Yoon图来比较吸附容量与平均Li+吸附速率(ASAR)之间的关系。与其他三种电极相比,LFP@C-7的吸附容量和ASAR皆最大。使用雷达图对不同样品的比容量、ASAR、容量保持率、D和Li+提取能力进行的综合评估,发现LFP@C-7的性能最佳,因此可用于后续研究。在1.00~1.60 V的截止电压范围内(不考虑水分解),进一步考察了LFP@C-7的Li+吸附容量和ASAR,可发现吸附容量和ASAR都随着截止电压的增加而逐渐增加,在1.6 V时达到了3.05mol g-1的最大吸附容量和0.1mmol g-1min-1的最大ASAR。
本研究侧重于选择性分离Li+,由于Mg2+的离子半径相似,因此被认为是最具干扰性的金属离子。为了评估LFP@C-7//AC在Mg2+/Li+混合溶液中分离Li+的能力,研究了不同Mg2+/Li+比例(1、2、10、30、40和60),发现LFP@C-7对Li+的选择性随着Mg2+/Li+增大出现先增加后降低的趋势,在比率为60时,选择性系数达到峰值212。这表明LFP@C-7具有极佳的Li+选择性,并具备从高Mg2+浓度的盐水中提取Li+的潜力。为了进一步评估选择性,使用了浓度为400 mg L-1,含有单一盐类(LiCl、KCl、CaCl2、MgCl2和NaCl)的溶液考察了不同离子在LFP@C-7//AC中的吸附/解吸行为。Li+的吸附速率比其他离子更快。Li+的最终吸附量(3.06 mmol g−1)明显高于Mg2+(1.06 mmol g−1)、Ca2+(0.89 mmol g−1)、K+(1.42 mmol g−1)和Na+(1.47 mmol g−1),这与比电容结果一致,表明 LFP@C-7具有优异的Li+选择性。
进一步探讨了LFP@C-7在模拟海水中的应用潜力,研究通过构建 LFP@C-7//AC系统,利用40 mL混合盐浓度为1000 mg L-1的模拟海水,进一步探索了LFP@C-7在Li+提取方面的应用潜力。结果发现电极对Li+的提取量高达3.05mmol g-1。相比之下,Ca2+、Mg2+和K+的提取量则偏低,Na+几乎可忽略不计。这些结果清楚地表明,LFP@C-7从多盐废水中高效提取Li+的潜力巨大。在本研究中,没有考虑去除聚磷菌衍生的生物碳,具体原因如下:首先,衍生碳作为一种还原剂,在防止LFP中的Fe2+氧化方面起着至关重要的作用。此外,衍生碳还能形成导电网络,增强LFP的导电性,改善LFP与Li+之间的接触。最后,衍生碳在抑制LFP聚集和缓解应力集中方面同样具有关键作用。
进一步评估了LFP@C-7相对于其他已报道电极的Li+提取性能,比较发现,LFP@C-7具有较高的容量和优异的循环稳定性。在使用1000mg L−1浓度的LiCl、1.60 V截止电压条件下进行的LFP@C-7长周期CDI实验中,可观察到电导率在不同循环中降低的大小波动较小。电极吸附容量在前5个循环期间出现波动,但在随后的循环中趋于稳定。100次循环后,容量保持率为83.3%,这表明LFP@C-7具有出色的循环和再生性能。LFP@C-7的SEC范围为2.54-3.40kWh mol-1,较文献比较偏低。LFP@C-7卓越的吸附和循环性能主要归功于其独特的结构及低反位缺陷的作用。具有低浓度反位缺陷的LFP@C-7形状如海胆,暴露出丰富的活性位点,而较薄的厚度和光滑的1D通道缩短了离子和电子的传输路径,从而显著提高了Li+的存储容量和速率。此外,LFP@C-7的高结晶度(83.2%)使其晶体结构稳定,能够承受Li+插入时产生的各向异性膨胀,因此可有效缓解相变引起的应力集中,从而不会出现颗粒粉化和主动失效。此外,含低浓度反位缺陷的LFP与聚磷菌衍生碳壳的复合,实现了EDL和赝电容耦合,并有助于缓解体积变化和防止LFP颗粒的聚集,从而显著提高了电极的循环稳定性。
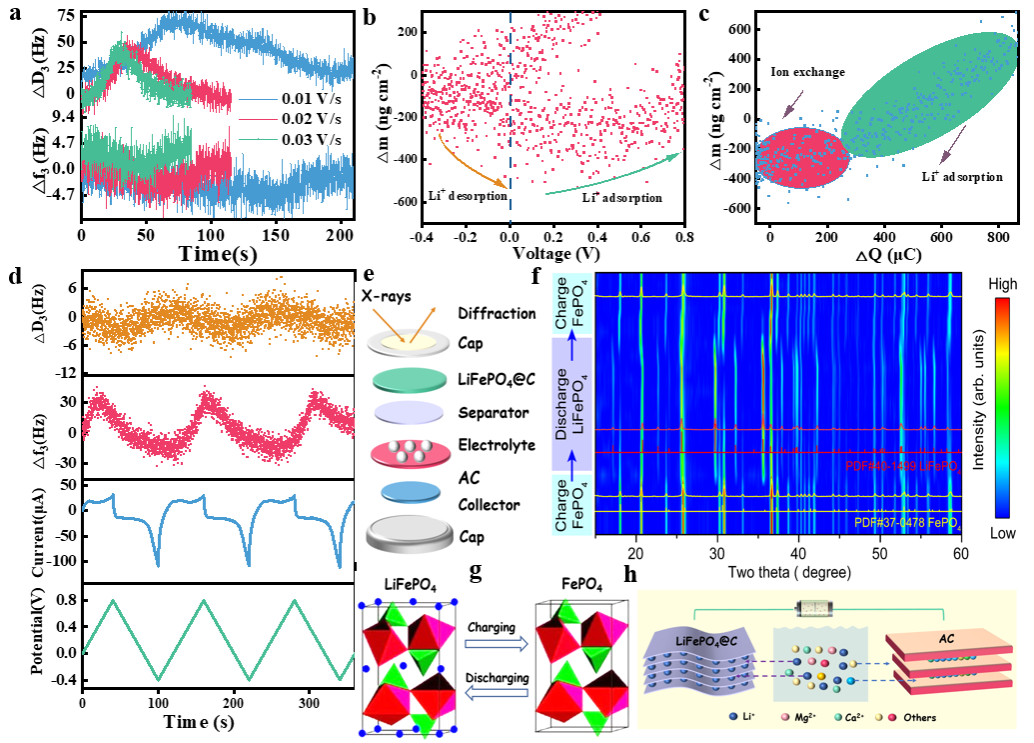
Figure 6.Mechanistic analysis.(a)Δf3/3 and ΔD3ofLFP@C-7 versus time at various scan rates during the 2nd CV cycle tested via EQCM.(b)The mass change of LFP@C-7 at the scan rate of 30 mV s-1.(c) LFP@C-7electrode mass versus charge passed during reduction at 30 mV s-1from EQCM-D.(d)Δf3andΔD3responses ofLFP@C-7from EQCM-D during CV at the scan rate of30 mV s-1in 0.01 MLiCl.(e)Schematic diagrams of the customized cell for in situ XRD measurements. (f) In situ XRD patterns of the LFP@C-7//AC cell during desalination/regeneration process. (g)Physical phase changes of LFP@C-7 during Li+embedding and removal processes. (h)Mechanism diagram of LFP@C-7//AC selective Li+extraction.
如图6所示,利用EQCM-D探讨了Li+从LFP@C-7电极中嵌入和移除的机制。LFP@C-7在0.01~0.03 V s-1下进行CV循环时,频率(Δf3/3)和耗散(ΔD3)的变化趋势相同。根据 EQCM-D 测试结果,进一步利用绍尔布雷方程量化LFP@C-7电极反应过程中的质量变化。结果表明,在阳极区间以0.02 V s-1扫描时,LFP@C-7电极的质量逐渐减小,当扫描到0 V时,质量变化达到最大值为217 ng cm-2,成功证实Li+在LFP@C-7电极中发生了嵌入和脱除过程。此外,根据质量变化,Li+分离过程可分为三个阶段。在第一阶段,H2O分子和Li+在LFP@C-7电极内共存,导致质量增加。在第二阶段,发生离子交换过程以建立吸附/解吸平衡,从而导致轻微的质量变化。在第三阶段,吸附的Li+迅速释放,电极质量恢复到初始值。
为了深入了解LFP@C-7电极的Li+捕获机制,使用定制腔室进行了原位XRD表征。图6f显示了LFP@C-7//AC体系在不同截止电压下的原位XRD图样,证实了CDI反应过程中离子的嵌入和脱嵌过程。根据对XRD特征峰变化的分析可知,在第一阶段的充电过程中,LiFePO4(PDF#40-199)转变为FePO4(PDF#37-0478),物理相变明显;在第二阶段的放电过程中,由于Li+的嵌入,FePO4转变为LFP;在第三阶段继续充电,得到与第一阶段相同的结果,即LFP 转化为FP,结合三阶段的原位XRD测试结果,可以推断LFP@C-7电极发生了FePO4→LiFePO4→FePO4的相变过程。因此,Li+的分离受到可逆插层和去插层的显著影响。LFP@C-7分离 Li+的实质是LFP和FP两相之间的转变( 图6.50 ),从而在多金属离子(Mg2+、Ca2+、K+等)存在时,优先实现Li+选择性地嵌入。
总结与展望
LFP作为Li+捕获阴极的一个关键障碍是开发经济的制造工艺。不同低温合成方法导致的反位缺陷(Li和Fe)仍是当前合成LFP的主要问题。LFP中的Li+运动是通过1D通道(沿b轴)进行的,反位缺陷不仅会阻碍Li+沿最快的扩散路径传输,而且还会引起额外的静电排斥,从而导致LFP晶体结构不稳定。因此,开发绿色经济并含有低浓度反位缺陷的LFP合成策略对于改善扩散动力学及循环稳定性具有重要意义。本章报道了一种利用微生物“代谢催化”功能介导合成含有低浓度反位缺陷LFP的策略。得到结论如下:
(1)基于聚磷菌自身所含酶类将摄入离子还原、转化、封盖和稳定的特性,实现了PO43-、Fe3+和Li+的“代谢催化”转化,成功合成了LFP@C。考察pH对合成产物形貌的影响,在不同pH条件下,LFP的形貌分别为绣球状(pH = 5)、手风琴(pH = 6)、海胆状(pH = 7)和海参状(pH = 8)。LFP@C-7的尺寸为50-100 nm,比表面积为200.8 m2g–1,LFP@C-7中LFP的含量约为83.2%。
(2)LFP@C-7具有优异的电化学性能,作为Li+选择性阴极时,LFP@C-7的Li+分离容量为3.05 mmol g-1,明显高于Ca2+(0.89 mmol g−1)和Mg2+(1.06 mmol g−1),当Mg2+/Li+比为60时,选择性系数达到峰值(212),表明其具有优异的Li+选择性能,具备从高Mg2+浓度盐水中分离Li+的潜力。
(3)XRD、拉曼、FTIR测试发现LFP@C含有低浓度反位缺陷(< 3%)和稳定的晶体形态。电化学测试发现LFP@C-7仅在LiCl溶液中出现充放电反应平台和氧化还原峰。EQCM-D测试发现Li⁺水合和去水化过程提高了Li⁺层间迁移速率并增加了活性面积,从而增强了Li⁺分离性能。原位XRD结果发现Li+脱嵌过程发生了FePO4→LiFePO4→FePO4的转化反应,可逆相变的循环有效提升了LFP@C-7的稳定性。
文献链接
https://doi.org/10.1016/j.nantod.2025.102781
另:封面图